The process of getting a drug to market, from first testing to final FDA approval, is summarized in figure 1 and described at greater length below.
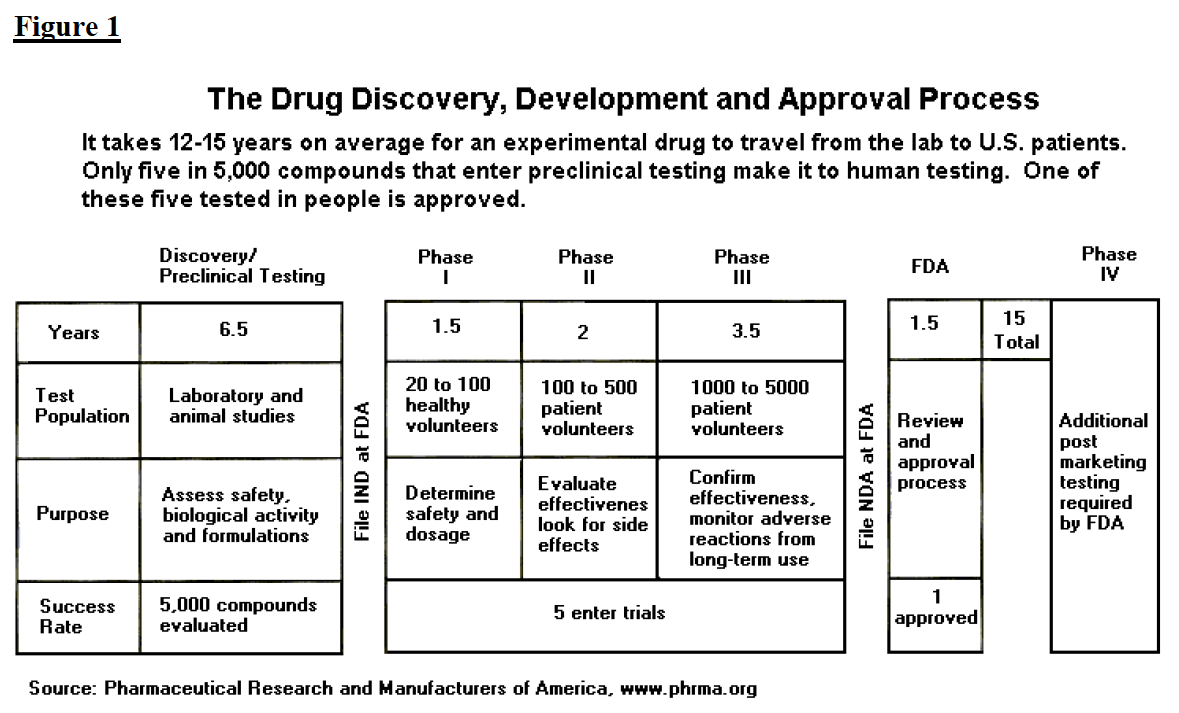
Drug companies continuously analyze thousands of compounds, seeking ones of therapeutic value. During the six to seven years of preclinical testing, the manufacturer completes synthesis and purification of the drug and conducts limited animal testing. Of five thousand compounds tested, approximately five will appear promising enough to induce the company to file an Investigational New Drug Application (IND). If the IND is approved by the FDA and by an Institutional Review Board, the manufacturer may begin the first phase of development.
The IND stage consists of three phases. In phase I, clinical trials using healthy individuals are conducted to determine the drug’s basic properties and safety profile in humans. Typically the drug remains in this stage for one to two years. In phase II, efficacy trials begin as the drug is administered to volunteers of the target population. At the end of phase II, the manufacturer meets with FDA officials to discuss the development process, continued human testing, any concerns the FDA may have, and the protocols for phase III, which is usually the most extensive and most expensive part of drug development. During the phases of the IND, the manufacturer can obtain accelerated development/review of the drug. Other accommodations for usage prior to approval include treatment IND and parallel tracking.
Once phase III is complete, the manufacturer files an NDA. Review of the NDA typically lasts one to two years, bringing total drug development and approval (that is, the IND and NDA stages) to approximately nine years. During the NDA stage, the FDA consults advisory committees made of experts to obtain a broader range of advice on drug safety, effectiveness, and labeling. Once approved, the drug may be marketed with FDA regulated labeling. The FDA also gathers safety information as the drug is used and adverse events are reported, and it will occasionally request changes in a labeling or will submit press releases as new contraindications arise. If adverse events appear to be systematic and serious, the FDA may withdraw a product from the market.
Over time there has been a clear tendency for FDA regulations and requirements to expand and multiply. In 1980, the typical drug underwent thirty clinical trials involving about fifteen hundred patients. By the mid-1990s, the typical drug had to undergo more than sixty clinical trials involving nearly five thousand patients.
Public Agenda Online has some useful background information on medical and drug research in the form of easy-to-read charts.